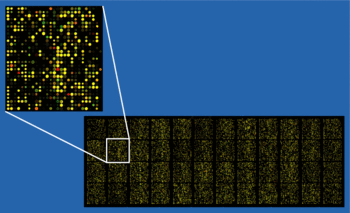
packageMicroarray;use5.006;usestrict;usewarnings;useExporter;our@ISA=qw( Exporter );our$VERSION='0.43';requireMicroarray::File;useMicroarray::Reporter;useMicroarray::Spot;requireMicroarray::File::Data;requireMicroarray::File::Data::Quantarray;requireMicroarray::File::Data::GenePix;requireMicroarray::File::Data::BlueFuse;# Constructorsubnew {my$class=shift;my$self= { };bless$self,$class;$self->barcode(shift);# barcode firstif(@_){# data file passedmy$data_file=shift;# data file$self->data_file($data_file);# then load the data_file object}return$self;}############################# general getter setters #############################subbarcode {my$self=shift;@_?$self->{ _barcode } =shift:$self->{ _barcode };}subdata_file {my$self=shift;if(@_) {my$data_file=shift;if((ref$data_file) && ($data_file->isa('data_file')) ){# if data_file object, load directly$self->{ _data_file } =$data_file;}else{# otherwise, if passed a file namemy$oData_File= data_file->new($data_file);# create the data_file object - let it guess the file format$self->{ _data_file } =$oData_File;# then load the data_file object}}else{$self->{ _data_file };}}# how are missing samples described in the GAL file?subblank_feature {my$self=shift;if(@_) {$self->{ _blank_feature } =shift;}else{if(defined$self->{ _blank_feature }) {$self->{ _blank_feature };}else{$self->default_blank_feature;}}}subdefault_blank_feature {'n/a';}# is there an experimental prefix to feature id?# anything beginning with a 'y' will be classed as 'yes', all others 'no'subprefix {my$self=shift;if(@_) {$self->{ _prefix } =shift;}else{if(defined$self->{ _prefix }) {$self->{ _prefix };}else{$self->default_prefix;}}}subdefault_prefix {'no'}subchannel1_dye_name {my$self=shift;my$data_file=$self->data_file;$data_file->channel1_name;}subchannel2_dye_name {my$self=shift;my$data_file=$self->data_file;$data_file->channel2_name;}sublong_ch1_name {my$self=shift;'ch1 ('.$self->channel1_dye_name.')';}sublong_ch2_name {my$self=shift;'ch2 ('.$self->channel2_dye_name.')';}subwhich_channel {my$self=shift;my$dye_name=shift;# assumes channels are different dyes!if($self->channel1_dye_name eq$dye_name) {return'ch1';}elsif($self->channel2_dye_name eq$dye_name) {return'ch2';}else{returnundef;}}# setter for data_file parameters used in feature selectionsubset_param {my$self=shift;my%hArgs=@_;while(my($arg,$val) =each%hArgs){if($self->can($arg)){$self->$arg($val);}else{die"Microrray ERROR; No parameter '$arg' is defined\n";}}}############################### Microarray reporter methods ################################ set reporters scrolls through the spot data# and fills a reporter object with all corresponding spot objectssubset_reporters {my$self=shift;$self->{ _reporters } = { };my$blank_feature=$self->blank_feature;# missing samplesmy$data_file=$self->data_file;$data_file->set_spot_objects;my$aSpots=$data_file->get_spots;SPOT:for(my$i=1;$i<@$aSpots;$i++) {my$oSpot=$$aSpots[$i];nextSPOTunless$oSpot;nextSPOTunless($oSpot->feature_id);nextSPOTif($oSpot->feature_id =~ /$blank_feature/i);$self->add_spot_to_reporter($oSpot);}}# uses the spot feature_id to determine the array reporter_idsubadd_spot_to_reporter {my$self=shift;my$oSpot=shift;if(my$oReporter=$self->get_reporter($oSpot->feature_id)) {# i.e. reporter already defined$oReporter->add_reporter_spot($oSpot);}else{# create a new reporter containing this spotmy$oReporter= array_reporter->new($oSpot->feature_id);$oReporter->add_reporter_spot($oSpot);$self->add_reporter($oReporter);}}subadd_reporter {my$self=shift;my$oReporter=shift;my$hReporters=$self->get_all_reporters;$hReporters->{$oReporter->reporter_id } =$oReporter;}subget_reporter {my$self=shift;my$reporter_id=shift;my$hReporters=$self->get_all_reporters;returnunless(defined$hReporters->{$reporter_id});$hReporters->{$reporter_id};}# returns a hash of reporters; key=reporter_id, value=reporter objectsubget_all_reporters {my$self=shift;unless(defined$self->{ _reporters }){$self->set_reporter_data;}$self->{ _reporters };}# returns an arrayref of spot objects for a given reporter_idsubget_reporter_spots {my$self=shift;my$oReporter=$self->get_reporter(shift);$oReporter->get_reporter_spots;}# returns an array of all reporter objectssubget_reporter_objects {my$self=shift;my$hReporters=$self->get_all_reporters;my@aValues=values%$hReporters;return\@aValues;}# returns an array of all reporter idssubget_reporter_ids {my$self=shift;my$hReporters=$self->get_all_reporters;my@aKeys=keys%$hReporters;return\@aKeys;}subset_reporter_data {my$self=shift;unless(defined$self->{ _reporters }){$self->set_reporters;}my$aReporters=$self->get_reporter_objects;formy$oReporter(@$aReporters) {$self->sort_reporter_data($oReporter);#$self->set_genetic_data($oReporter);}}subsort_reporter_data {my$self=shift;my$oReporter=shift;my$aSpots=$oReporter->get_reporter_spots;# setting these variables now saves making many calls to the same methods!my$hBad_Flags=$self->data_file->bad_flags;my$low_signal=$self->low_signal;my$high_signal=$self->high_signal;my$percen_sat=$self->percen_sat;my$min_snr=$self->min_snr;my$signal_quality=$self->signal_quality;my$min_diameter=$self->min_diameter;my$max_diameter=$self->max_diameter;# my $max_pixels = $self->max_pixels;# my $min_pixels = $self->min_pixels;SPOT:formy$oSpot(@$aSpots) {$oSpot->spot_status(0);# set spot to 'rejected' at startnextSPOTif(defined$hBad_Flags->{$oSpot->flag_id });unless($self->should_ignore_signal_qa){######## SIGNAL QUALITY ASSESSMENTS ########if(($oSpot->channel1_signal <$low_signal) ||($oSpot->channel1_signal >$high_signal) ||($oSpot->channel1_sat && ($oSpot->channel1_sat >$percen_sat)) ||($oSpot->channel1_snr <$min_snr) ||($oSpot->channel1_quality <$signal_quality) ||($oSpot->channel2_signal <$low_signal) ||($oSpot->channel2_signal >$high_signal) ||($oSpot->channel2_sat && ($oSpot->channel2_sat >$percen_sat)) ||($oSpot->channel2_snr <$min_snr) ||($oSpot->channel2_quality <$signal_quality) ){nextSPOT;}}unless($self->should_ignore_spot_qa){######## SPOT QUALITY ASSESSMENTS ########if(($oSpot->spot_diameter <$min_diameter) ||($oSpot->spot_diameter >$max_diameter)){nextSPOT;}}# spot passes quality assessment$oSpot->spot_status(1);$oReporter->good_spot;# for calculation of modal signal ratios$self->all_ch1($oSpot->channel1_signal);$self->all_ch2($oSpot->channel2_signal);# for calculation of feature signal ratios$oReporter->all_ch1($oSpot->channel1_signal);$oReporter->all_ch2($oSpot->channel2_signal);# for some plots$self->x_pos($oSpot->x_pos);$self->y_pos($oSpot->y_pos);unless($oSpot->channel2_signal == 0){$self->all_ratios(($oSpot->channel1_signal)/($oSpot->channel2_signal));$oReporter->all_ratios(($oSpot->channel1_signal)/($oSpot->channel2_signal));}}}# some ideas for filtering using environment variables# sub filter_or_not {# my $self = shift;# if (@_){# if (shift eq 'Y'){# $ENV{ FILTER } = 'Y';# } else {# $ENV{ FILTER } = 'N';# }# }# }# sub {# $ENV{ REJECT_UNIQUE }# }# sub filter_values {# $ENV{ FILTER_VALUES } = [500,0.5];# }# sub filter_on {# $ENV{ FILTER_ON } = [channel_signal,channel_quality];# }subignore_signal_qa {my$self=shift;$self->{ _ignore_signal_qa }++;}subignore_spot_qa {my$self=shift;$self->{ _ignore_spot_qa }++;}subshould_ignore_signal_qa {my$self=shift;$self->{ _ignore_signal_qa };}subshould_ignore_spot_qa {my$self=shift;$self->{ _ignore_spot_qa };}# the methods all_ch1/ch2/ratios create an array ref# containing all the relevant values from the array# these arrayrefs can be analysed for QC purposes# using Statistics::Descriptivesuball_ch1 {my$self=shift;unless(defined$self->{ _all_ch1 }){$self->{ _all_ch1 } = [];}if(@_){my$aCh1_Signals=$self->{ _all_ch1 };push(@$aCh1_Signals,shift);}else{$self->{ _all_ch1 };}}suball_ch2 {my$self=shift;unless(defined$self->{ _all_ch2 }){$self->{ _all_ch2 } = [];}if(@_){my$aCh2_Signals=$self->{ _all_ch2 };push(@$aCh2_Signals,shift);}else{$self->{ _all_ch2 };}}subx_pos {my$self=shift;unless(defined$self->{ _x_pos }){$self->{ _x_pos } = [];}if(@_){my$aX_Pos=$self->{ _x_pos };push(@$aX_Pos,shift);}else{$self->{ _x_pos };}}suby_pos {my$self=shift;unless(defined$self->{ _y_pos }){$self->{ _y_pos } = [];}if(@_){my$aY_Pos=$self->{ _y_pos };push(@$aY_Pos,shift);}else{$self->{ _y_pos };}}suball_ratios {my$self=shift;unless(defined$self->{ _all_ratios }){$self->{ _all_ratios } = [];}if(@_){my$aRatios=$self->{ _all_ratios };push(@$aRatios,shift);}else{$self->{ _all_ratios };}}# summary of why spots were rejectedsuberror_report {my$self=shift;if(defined$self->{ _error_report }) {$self->{ _error_report };}else{$self->{ _error_report } = { };}}################################## Getter setters for spot ## quality assessment criteria ################################### signal levels; set to linear range of the scannersublow_signal {my$self=shift;if(@_) {$self->{ _low_signal } =shift;}else{if(defined$self->{ _low_signal }) {$self->{ _low_signal };}else{$self->default_low_signal;}}}subdefault_low_signal {5000;}subhigh_signal {my$self=shift;if(@_) {$self->{ _high_signal } =shift;}else{if(defined$self->{ _high_signal }) {$self->{ _high_signal };}else{$self->default_high_signal;}}}subdefault_high_signal {60000;}# % of pixels that are saturated# provides check that signals are within the linear range# and also helps to flag 'dirty' spotssubpercen_sat {my$self=shift;if(@_) {$self->{ _percen_sat } =shift;}else{if(defined$self->{ _percen_sat }) {$self->{ _percen_sat };}else{$self->default_percen_sat;}}}subdefault_percen_sat {10;}# minimum acceptable spot signal:noise ratiosubmin_snr {my$self=shift;if(@_) {$self->{ _snr } =shift;}else{if(defined$self->{ _snr }) {$self->{ _snr };}else{$self->default_min_snr;}}}subdefault_min_snr {10;}# subjective assessment of signal quality, using (% signal > B + 2SD) or bluefuse's confidence valuesubsignal_quality {my$self=shift;if(@_) {$self->{ _signal_quality } =shift;}else{if(defined$self->{ _signal_quality }) {$self->{ _signal_quality };}else{$self->default_signal_quality;}}}subdefault_signal_quality {95;}# spot size#Êby combining stringent diameter checking with#Êexpected pixel number, we can check the# 'circularity' of a spotsubmin_diameter {my$self=shift;if(@_) {$self->{ _min_diameter } =shift;}else{if(defined$self->{ _min_diameter }) {$self->{ _min_diameter };}else{$self->default_min_diameter;}}}subdefault_min_diameter {80;}submax_diameter {my$self=shift;if(@_) {$self->{ _max_diameter } =shift;}else{if(defined$self->{ _max_diameter }) {$self->{ _max_diameter };}else{$self->default_max_diameter;}}}subdefault_max_diameter {150;}subtarget_diameter {my$self=shift;if(@_) {$self->{ _target_diameter } =shift;}else{if(defined$self->{ _target_diameter }) {$self->{ _target_diameter };}else{$self->default_target_diameter;}}}subdefault_target_diameter {100;}submax_diameter_deviation {my$self=shift;if(@_) {$self->{ _max_diameter_deviation } =shift;}else{if(defined$self->{ _max_diameter_deviation }) {$self->{ _max_diameter_deviation };}else{$self->default_max_diameter_deviation;}}}subdefault_max_diameter_deviation {10;}submin_pixels {my$self=shift;if(@_) {$self->{ _min_pixels } =shift;}else{unless(defined$self->{ _min_pixels }) {$self->{ _min_pixels } =$self->pixel_area($self->min_diameter);}$self->{ _min_pixels };}}submax_pixels {my$self=shift;if(@_) {$self->{ _max_pixels } =shift;}else{unless(defined$self->{ _max_pixels }) {$self->{ _max_pixels } =$self->pixel_area($self->max_diameter);}$self->{ _max_pixels };}}subpixel_area {my$self=shift;my$micron_diameter=shift;# spot diameter in micronsmy$data_file=$self->data_file;my$pixel_size=$data_file->pixel_size;# each channel, in micronsmy$pixel_radius= ($micron_diameter/$pixel_size)/2;# radius as number of pixelsmy$pixel_area= 3.14159 * ($pixel_radius*$pixel_radius);# area as number of pixelsreturnint($pixel_area+ 0.49999);# ensure correct rounding of a positive value}subtarget_pixels {my$self=shift;if(@_) {$self->{ _target_pixels } =shift;}else{if(defined$self->{ _target_pixels }) {$self->{ _target_pixels };}else{$self->pixel_area($self->default_target_diameter);}}}subnormalisation {# modal log2 ratio normalisationmy$self=shift;if(@_){$self->{ _normalisation } =shift;}else{unless(defined$self->{ _normalisation }){return'yes';}if($self->{ _normalisation } =~ /^n/i){returnundef;}else{return'yes';}}}subsignal_normalisation {# from scanner outputmy$self=shift;if(@_){$self->{ _signal_normalisation } =shift;}else{unless(defined$self->{ _signal_normalisation }){return'yes';}if($self->{ _signal_normalisation } =~ /^n/i){returnundef;}else{return'yes';}}}#Êgenetic_data_source defines whether we get the genetic data from file or database# currently, 'data_file' means from the results file (ie what was in the GAL file)# 'database' from array_pipeline_v4.chori_bac_clone_info# can expand to fetch it from separate file, and also to specify database table if differentsubgenetic_data_source {my$self=shift;if(@_) {$self->{ _genetic_data_source } =shift;}else{if(defined$self->{ _genetic_data_source }) {$self->{ _genetic_data_source };}else{$self->default_gendata_source;}}}subdefault_gendata_source {'data_file';}# user defined headers for data outputsubformat_headers {my$self=shift;if(@_){my@aHeaders=@_;$self->{ _format_headers } = \@aHeaders;}else{if(defined$self->{ _format_headers }){$self->{ _format_headers };}else{$self->default_format_headers;}}}### image output ###subset_image_data {my$self=shift;my$oImage=shift;$oImage->{ _ch1_values } =$self->all_ch1;$oImage->{ _ch2_values } =$self->all_ch2;$oImage->{ _x_coords } =$self->x_pos;$oImage->{ _y_coords } =$self->y_pos;$oImage->parse_args(@_);$oImage->process_data;# by-pass $oImage->set_data## ma/ri/scatter use _ch1_values/_ch2_values# heatmaps use ch1_values and x/y_coords## currently no support for direct plotting of cgh_plots# $self->{ _x_values } = $oData_File->all_locns;# this is the problem - don't yet have genomic locations being returned directly# should make a Microarray::CGH object and add this function# but how to easily integrate database support?# what about have a database object, which would be ensembl by default, but which will handle LIMS database# $self->{ _y_values } = $oData_File->all_log2_ratio;# $self->all_ratios;# $self->{ _reporter_names } = $oData_File->all_feature_names;# $self->get_reporter_ids;# $self->{ _cgh_calls } = $oData_File->cgh_calls if $oData_File->isa('cgh_call_output');###}subplot_ma {my$self=shift;my$oImage= ma_plot->new();$self->set_image_data($oImage,@_);$oImage->make_plot;}subplot_ri {my$self=shift;my$oImage= ri_plot->new();$self->set_image_data($oImage,@_);$oImage->make_plot;}subplot_intensity_scatter {my$self=shift;my$oImage= intensity_scatter->new();$self->set_image_data($oImage,@_);$oImage->make_plot;}subplot_log2_heatmap {my$self=shift;my$oImage= log2_heatmap->new();$self->set_image_data($oImage,@_);$oImage->make_plot;}subplot_intensity_heatmap {my$self=shift;my$oImage= intensity_heatmap->new();$self->set_image_data($oImage,@_);$oImage->make_plot;}}1;__END__=head1 NAMEMicroarray - A Perl module for creating and manipulating DNA Microarray experiment objects=head1 SYNOPSISuse Microarray;my $oArray = microarray->new($barcode,$data_file);$oArray->set_reporter_data;=head1 DESCRIPTION=begin html<style type="text/css">.thumb {margin-bottom: 0.5em;border-style: solid;border-color: white;width: auto;overflow: hidden;}.tright {float: right;clear: right;border-width: 0.5em 0 0.8em 1.4em;}.thumbinner {border: 1px solid #ccc;padding: 3px !important;background-color: #f9f9f9;font-size: 94%;text-align: center;overflow: hidden;}.thumbimage {border: 1px solid #ccc;}.thumbcaption {border: none;text-align: left;line-height: 1.4em;padding: 3px !important;font-size: 90%;font-style: italic;}</style><div class="thumb tright"><div class="thumbinner" style="width:352px;"><img src="http://upload.wikimedia.org/wikipedia/commons/thumb/0/0e/Microarray2.gif/350px-Microarray2.gif" width="350" height="213" class="thumbimage" alt="Example of an approximately 40,000 probe spotted oligo microarray with enlarged inset to show detail"><div class="thumbcaption">Example of an approximately 40,000 probe spotted oligo microarray with enlarged inset to show detail</div></div></div>=end htmlDNA Microarrays (L<http://en.wikipedia.org/wiki/Dna_microarray>) also known as 'Gene Chips' or 'DNA chips', are an experimental tool used in genetic research and other related disciplines. They consist of thousands of DNA probes immobilised on a solid surface (such as a glass slide) and enable high-resolution, high-throughput analyses of a variety of parameters such as gene expression, genetic variation, or chromosome copy number variants.=head2 TypicallyA single Microarray experiment (typically) generates large quantities of data which (typically) requires some form of post-processing before the data can be interpreted or visualised. The processing of microarray data is (typically) handled by a Bioinformatician (L<http://en.wikipedia.org/wiki/Bioinformatics>), and the favourite computer programming language of a Bioinformatician is (typically) Perl. However, until now the poor Bioinformatician has (typically) had to use a statistical programming language like R (L<http://www.r-project.org>) - not because it is intrinsically better for the job than Perl, but rather because there were no CPAN modules that helped the Bioinformatician to perform these tasks lazily, impatiently and with hubris.Microarray is a suite of object-oriented Perl Modules for the analysis of microarray experiment data. These include modules for handling common formats of microarray files, modules for the programmatic abstraction of a microarray experiment, and for the output of a variety of images describing microarray experiment data. Hopefully, this suite of modules will help Bioinformaticians to (typically) handle their data with laziness, impatience and hubris.=head2 How it worksThe Microarray object contains several levels of microarray associated data, organised in a (fairly) intuitive way. First, there's the data that you have obtained from a microarray scanner, in the form of a data file. This is imported into Microrray as a L<Data_File|Microarray::File::Data> object. Support for different data file formats is built into the L<Data_File|Microarray::File::Data> class, and creating new classes for your favourite scanner/software output is relatively simple. Data extracted from the microarray spots are then imported into individual L<array_spot|Microarray::Spot> objects. Next, replicate spots are collated into L<array_reporter|Microarray::Reporter> objects. Most of the quality control functions operating on parameters such as signal intensity and spot size, are built into this final process, so that an L<array_reporter|Microarray::Reporter> object only contains data from spots that have passed the QC assessments. Post-processing of the data is then performed using the L<Microarray::Analysis|Microarray::Analysis> module, and finally the data are visualised using the L<Microarray::Image|Microarray::Image> module.=head1 METHODS=head2 Creating microarray objectsThe microarray object is created by providing a barcode (or name) and a data file. It is assumed the data file contains minimal information about the reporter identities (i.e. name or id). In the case of a CGH-microarray, that means the BAC clone name/synonym at each spot. For cDNA or oligo arrays, that would mean a gene name, cDNA accession, or oligo name. Most of the functions between initialising the objects and returning formatted data can be accessed, and default settings can be changed (see below).=head2 Data FileThe data file can be passed to Microarray either as a file name, filehandle object, or L<data_file|Microarray::File::Data> object. If a filehandle is passed, the filename also needs to be set.$oArray = microarray->new($barcode,'my_file'); # will try to guess the file formator$oData_File = quantarray_file->new('my_file'); # create the data file...$oData_File = quantarray_file->new('my_file',$Fh); # can pass a filename and filehandle to the data file$oArray = microarray->new($barcode,$oData_File); # ...then load into microarray=head2 Feature Identification=over=item B<blank_feature>Defines how 'empty' spots are described in the data file. Default 'n/a'=item B<prefix>Set to 'y' if the feature id is prefixed in some way (for instance, we use prefixes to distinguish different methods used to prepare the same sample for microarray spotting). Default 'n'=back=head2 Changing Default SettingsThere are many parameters that are used in the process of defining features, and for their quality control. Below is an overview of the methods used. As well as being able to set these parameters individually, you can also set a number in one call using the set_param() method$oArray->set_param(min_diameter=>100,min_snr=>10);=head3 Spot Quality ControlThere are various (mostly self-explanatory) methods for setting spot quality control measurements, listed below=over=item B<low_signal>, B<high_signal>Defaults = 5000, 60000=item B<min_diameter>, B<max_diameter>Default = 80, 150=item B<min_pixels>Default = 80=item B<signal_quality>Varies depending on the data file format used; for the ScanArray format, this refers to the percentage of spot pixels that are more than 2 standard deviations above the background (default = 95); for BlueFuse this corresponds to the spot confidence value.=item B<percen_sat>The method percen_sat() refers to the percentage of spot pixels that have a saturated signal. Default = 10. Not relevant to BlueFuse format.=back=head3 Reporter Analysis=over=item B<normalisation>Set to either 'y' or 'n', to include ratio normalisation. Note: this is only base-level normalisation, not signal normalisation. For CGH-microarrays, this is a subtraction of the modal log2 ratio. Default = 'y'=back=head2 Access to Spot DataAll of the microarray data can be independently accessed in one of two ways. First, data can be obtained directly from the data file object, and in fact you could use this module just to simplify the data input process for your own applications and not use any of the other functions of Microarray. Individual spot objects can be returned by referring to their spot index (which is usually also the order they appear in the data file) or all spot objects can be returned as a list. See L<Microarray::Spot|Microarray::Spot> and L<Microarray::Reporter|Microarray::Reporter> for more information.my $oSpot = $oData_File->get_spots(1);my $aAll_Spots = $oData_File->get_spots;=head3 Data file methods=over=item B<file_name>Depending how you used Data_File, will be the name or the full path you provided=item B<get_header_info>For example in the ScanArray format, the data header contains information about the scan, such as laser power, PMT, etc=back=head2 Access to Reporter DataAlternatively you can access the reporter data, which collates replicate spot data. Either, individual reporter objects can be returned, and array_reporter methods applied to them, or all reporter objects/ids can be returned as a list.$oReporter = $oArray->get_reporter('reporter1'); # returns a single reporter object$aReporter_Objects = $oArray->get_reporter_objects; # returns a list of reporter objects$aReporter_Names = $oArray->get_reporter_ids; # returns a list of reporter ids$hReporters = $oArray->get_all_reporters; # returns a hash of reporters; key=reporter_id, value=reporter object=head2 Image OutputMicroarray will output QC/QA plots of the data as PNG files, using the L<Microarray::Image::QC_Plots|Microarray::Image::QC_Plots> module. Simply call any of the following methods to create the relevant plot, passing any plot parameters if required.my $plot_png = $oArray->plot_ma(scale=>50);open (PLOT,'>plot.png');print PLOT $plot_png;close PLOT;=over=item B<plot_ma>, B<plot_ri>Plots an MA/RI plot. (These are the same plot, just with different units).=item B<plot_intensity_scatter>A simple intensity scatter of channel1 signal vs channel2 signal.=item B<plot_log2_heatmap>A spatial plot of the log2 values from each spot of the array.=item B<plot_intensity_heatmap>A spatial plot of the signal intensity of each spot of the array.=back=head1 FUTURE DEVELOPMENTThis module is under continued development for our laboratory's microarray facility. If you would like to contribute to the development of Microarray, whether to add more advanced features of data analysis, or simply to add support for other microarray platforms/scanners, please contact the author.=head1 SEE ALSOL<Microarray::File|Microarray::File>, L<Microarray::Reporter|Microarray::Reporter>, L<Microarray::Spot|Microarray::Spot>=head1 AUTHORChristopher Jones, Translational Research Laboratories, Institute for Women's Health, University College London.c.jones@ucl.ac.uk=head1 COPYRIGHT AND LICENSECopyright 2007 by Christopher Jones, University College LondonThis library is free software; you can redistribute it and/or modifyit under the same terms as Perl itself.=cut
{kind=link}