NAME
FASTX::Abi - Read Sanger trace file (chromatograms) in FASTQ format. For traces called with hetero option, the ambiguities will be split into two sequences to allow usage from NGS tools that usually do not understand IUPAC ambiguities.
VERSION
version 1.0.1
SYNOPSIS
use FASTX::Abi;
my $trace_fastq = FASTX::Abi->new({ filename => '/path/to/trace.ab1' });
# Print chromatogram as FASTQ (will print two sequences if there are ambiguities)
print $trace_fastq->get_fastq();BUILD STATUS

The source from GitHub is tested using Travis-CI for continuous integration. Check also the CPAN grid test for a better estimate of build success using CPAN version of interest.
INSTALLATION
Via cpanminus:
# Install cpanminus if you don't have it:
curl -L https://cpanmin.us | perl - --sudo App::cpanminus
# Install FASTX::Abi
cpanm FASTX::AbiVia Miniconda https://docs.conda.io/en/latest/miniconda.html:
conda install -y -c bioconda perl-fastx-abiHETERO CALLING (IUPAC AMBIGUITIES)
When Sanger-sequencing a mix of molecules (i.e. PCR product from heterozigous genome) containing a single-base polimorphisms, if the .ab1 file is called using the hetero modality the sequence stored in the file will contain ambiguous bases (i.e. using DNA IUPAC characters).
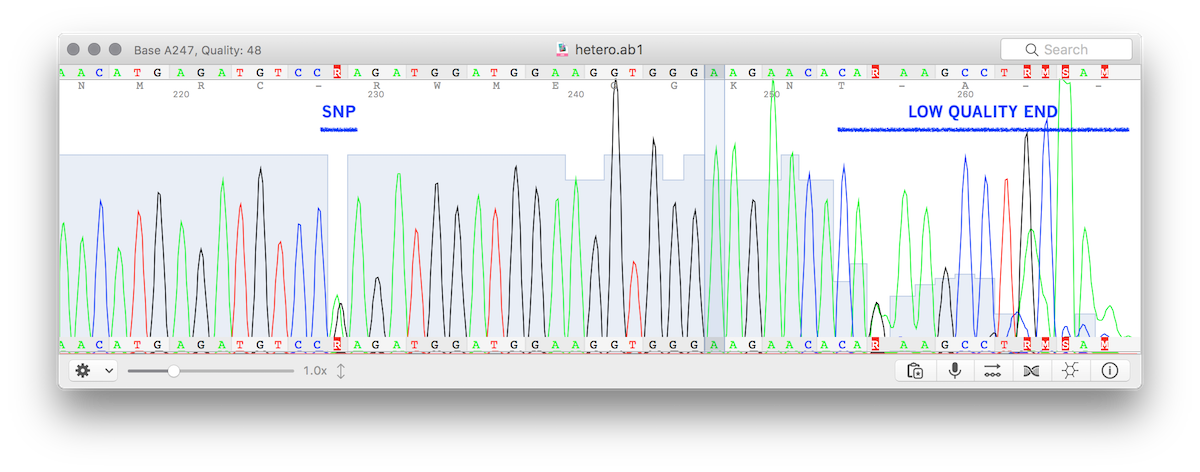
This module is designed to produce NGS-compatible FASTQ, so when ambiguous bases are detected the two "alleles" will be split into two sequences (of course, if more SNPs are present in the same trace, the output cannot be phased). The image below shows a trace file (.ab1) with a valid variant and a low quality end.
METHODS
new()
When creating a new object the only required argument is filename.
# Trimming is based on Bio::Trace::ABIF->clear_range()
my $trace_fastq = FASTX::Abi->new({
filename => "$filepath",
min_qual => 22,
wnd => 16,
bad_bases => 2,
keep_abi => 1, # keep Bio::Trace::ABIF object in $self->{chromas} after use
});
# Raw sequence and quality:
print "Raw seq/qual: ", $trace_fastq->{raw_sequence}, ", ", $trace_fastq->{raw_quality}, "\n";
# Trimmed sequence and quality:
print "Seq/qual: ", $trace_fastq->{sequence}, ", ", $trace_fastq->{quality}, "\n";
# If there are ambiguities (hetero bases, IUPAC):
if ($trace_fastq->{diff} > 0 ) {
print "Differences: ", join(',', @{ $trace_fastq->{diffs} }), "\n";
print "Seq 'A': ", $trace_fastq->{seq1}, "\n";
print "Seq 'B': ", $trace_fastq->{seq2}, "\n";
}Input parameters:
- filename, path
-
Name of the trace file (AB1 format)
- trim_ends, bool
-
Trim low quality ends (true by default, highly recommended)
- min_qual, int
-
Minimum quality value for trimming
- wnd, int
-
Window size for end trimming
- bad_bases, int
-
Maximum number of bad bases per window
get_fastq($sequence_name, $fixed_quality)
Return a string with the FASTQ formatted sequence (if no ambiguities) or two sequences (if at least one ambiguity is found).
If no $sequence_name is provided, the header will be made from the AB1 filename. If $sequence_name is defined and contains spaces, they will converted to underscores.
The $fixed_quality is a user provided fixed quality value for each base printed. Can be an integer (10 < x < 40), or a single char. In the first case it will be encoded as quality score (values above 93 will all be rendered as ~), in the second case the character will be used as quality score. If not supplied the original quality of the chromatogram will be used (that will be very low in SNPs positions).
# Use 40 as quality for each base of the trace:
$trace->get_fastq(undef, 40);get_trace_info()
Returns an object with trace information:
my $info = FASTX::Abi->get_trace_info();
print "Instrument: ", $info->{instrument}, "\n";
print "Version: ", $info->{version}, "\n";
print "Average peak distance: ", $info->{avg_peak_spacing}, "\n";rc()
$trace->rc();Reverse complement the chromatogram sequence.
merge()
Merge two chromatograms if overlapping returning the merged sequence.
my $merged = $trace->merge($other_trace);_get_sequence()
Internal routine (called by new()) to populate sequence and quality. See new()
SEE ALSO
This module is a wrapper around Bio::Trace::ABIF by Nicola Vitacolonna.
AUTHOR
Andrea Telatin <andrea@telatin.com>
COPYRIGHT AND LICENSE
This software is Copyright (c) 2019 by Andrea Telatin.
This is free software, licensed under:
The MIT (X11) License